 Neurofibromatosis (NF) is a condition that causes tumors to grow on nerve tissue, producing skin and bone abnormalities.
Neurofibromatosis (NF) is a condition that causes tumors to grow on nerve tissue, producing skin and bone abnormalities. NF is often diagnosed in childhood, occasionally in infancy (in children with severe cases), but usually around 3-16 years of age. Effects of the disease vary widely — some children live almost unaffected by the condition; rarely, others might be severely disabled.
There's no specific cure for NF, but tumors usually can be removed and related complications treated. Because learning disabilities occur in about half the children with NF, some might need extra help in the classroom.
About Neurofibromatosis
Neurofibromatosis is a neurocutaneous syndrome passed down through the parents' genes, and it affects the brain, spinal cord, nerves, skin, and other systems in the body.
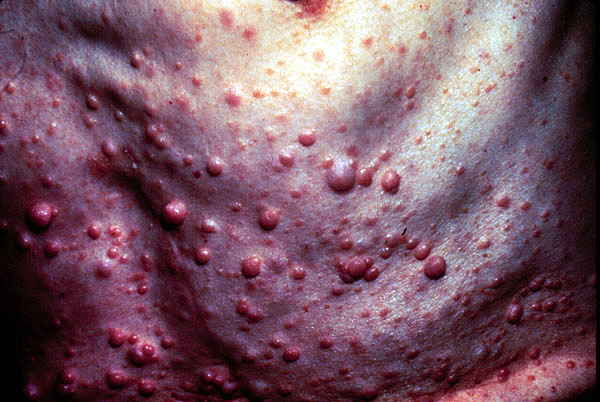
Neurofibromatosis is defined by tumors, called neurofibromas, that grow along nerves in the body, or on or under the skin. As the tumors increase in size, they can press on vital areas of the body, causing problems in the way the body functions.
Neurofibromas often first appear in childhood, especially during puberty. The first noticeable sign is almost always the presence of brown café au lait spots. These distinctive spots don't hurt or itch and never progress to anything more serious than spots. They can be found anywhere on the body, though not usually on the face. Tiny ones — freckles — may be seen under the arms or in the groin area.
Many neurofibromas can be removed. Although usually benign (noncancerous), an estimated 3%-5% become cancerous.
Of the two types of neurofibromatosis — NF1 and NF2 — NF1 is more common, occurring in 1 of every 4,000 births and affecting an estimated 100,000 Americans. It is also known as von Recklinghausen disease.
NF2 is characterized by the presence of bilateral acoustic neurofibroma-like tumors and is rarer, seen in 1 in 50,000 births. People with NF2 usually develop benign tumors on the nerves in their ears, causing hearing loss, eventual deafness, and problems with balance.
The severity of both types of neurofibromatosis varies greatly. In families where more than one person has NF, it can present with different physical signs and complications for each person. At diagnosis, it isn't possible to know right away whether a case will be mild or lead to severe complications.
Causes of NF
Both types of neurofibromatosis are autosomal dominant genetic disorders, which means an affected person has 1 chance in 2 of passing it on with each pregnancy. Neurofibromatosis also can be the result of a spontaneous change (mutation) in the genetic material of the sperm or egg at conception in families with no previous history of NF. About half of cases are inherited, and the other half are due to spontaneous genetic mutation.
NF 1 and NF 2 are each related to changes in separate genes. The NF1 gene is located on chromosome 17, and NF2 has been traced to chromosome 22. These findings are important because they may eventually lead to the development of a blood test or other genetic test to find out if a relative has NF.
Signs and Symptoms
NF1 is sometimes diagnosed in younger children, especially those with more severe forms of the disorder. One key to early diagnosis of mild NF is the appearance of café-au-lait spots on the skin.
Many people who do not have NF have a few café-au-lait spots. But if a young child has five or more, at least ½ inch in size (roughly the size of a dime), a doctor will look for other clues that may indicate NF, including neurofibromas — tumors on, under, or hanging off the skin — and Lisch nodules, tiny, noncancerous tumors on the iris (the colored part of the eye). Lisch nodules are of no clinical significance except that they help confirm a diagnosis of NF.
Neurofibromas often become evident on various parts of the body, beginning at the arms, around 10 years of age. A child may also develop freckling in the folds of the skin of the armpit or groin or on other parts of the body where the skin creases.
Abnormalities of the skeleton, such as the thinning or overgrowth of the bones in the arms or lower leg, curvature of the spine (scoliosis), and other bone deformities also may be features of NF1.
NF2 is usually not diagnosed until a child is older. Hearing loss in the late teens and early twenties is often among the first symptoms of the disorder, and is caused by tumors growing on the auditory nerves (which carry electrical impulses from the inner ear to the brain, allowing us to hear) on one or both sides.
Other symptoms of NF2 include continuous ringing in the ears, headache, facial pain or weakness, and feeling unsteady or off balance.
Diagnosis and Treatment
Neurofibromatosis is usually diagnosed based on a combination of findings. A child must have at least two of the following signs to be diagnosed with NF1:
- café-au-lait spots of a certain number, size, and location
- the appearance of two or more neurofibromas (often resembling pea-sized bumps on the skin)
- Lisch nodules on the irises
- an optic glioma (tumor along the main nerve of the eye that is responsible for sight)
- certain skeletal abnormalities
- a family member with NF1
- freckling under the arms or in the groin
Tests like magnetic resonance imaging (MRI) and X-rays may be used to screen for tumors or evidence of skeletal problems. A child's head circumference will be measured, as kids with symptoms of NF can have a circumference that's larger than normal for their age. Blood pressure will be monitored. Doctors also take a detailed personal history, looking for signs of learning difficulties.
To diagnose NF2, doctors will check for any evidence of hearing loss. They'll order audiometry (hearing tests) as well as imaging tests to look for tumors in the nerves of the ears, spinal cord, or brain. They'll also determine if there's a family history of NF2.
Genetic testing is now available for people with a family history of either NF1 or NF2, though such testing is still not 100% sensitive. Amniocentesis or chorionic villus sampling can sometimes determine if an unborn child has the condition.
Treatment for NF1 includes removal of the neurofibromas for cosmetic purposes, treating the complications (see below), and getting intervention for children with learning disabilities. Kids will be referred to appropriate medical specialists to monitor and treat complications, which may include:
- seizures (up to 40% of children with NF1 have them)
- high blood pressure
- scoliosis
- speech impairment
- optic nerve tumors (which can cause vision problems leading to blindness)
- early or delayed onset of puberty
Rarely, neurofibromas can become cancerous (3%-5% of cases). In these occurrences, surgery, chemotherapy, or radiation may be necessary.
With NF2, surgeons will likely need to remove the auditory nerve tumors, which may cause deafness afterward. When parts of the auditory nerve are removed, hearing aids won't work.
In 2000, the U.S. Food and Drug Administration (FDA) approved an auditory brainstem implant for people with NF2 who have lost their hearing. This device transmits sound signals directly to the brain, enabling the person to hear certain sounds and speech.
Currently, researchers are conducting trials with medications in the hopes they'll be able to offer more treatment options.
Caring for Your Child
The first noticeable sign of neurofibromatosis usually is the presence of multiple café-au-lait spots. If your child has several of these spots, ask your doctor to do a thorough examination; he or she may need to screen your child for other signs of NF.
If your child has already been diagnosed with NF and you notice that a growing tumor is beginning to cause a problem, tell your doctor immediately.
One of the most important things you can do is get early intervention if your child has learning disabilities. It also helps to seek out support groups that can provide your family with practical advice and encouragement.
Remember, most people (about 60%) diagnosed with NF1 have only relatively mild signs of the disorder, like café-au-lait spots and a few neurofibromas on the surface of the skin, which require little or no treatment.
Kids diagnosed with mild NF who remain fairly healthy into early adulthood are less likely to develop more serious complications later in life. Kids diagnosed with more serious forms often have correctable complications and with appropriate help and support can lead happy and productive lives.
 Neurocutaneous syndromes are disorders that lead to abnormal growth of tumors in various parts of the body. They're caused by the abnormal development of cells in an embryo and characterized by the presence of tumors in various parts of the body (including the nervous system) and by certain differences in the skin.
Neurocutaneous syndromes are disorders that lead to abnormal growth of tumors in various parts of the body. They're caused by the abnormal development of cells in an embryo and characterized by the presence of tumors in various parts of the body (including the nervous system) and by certain differences in the skin.  Muscular dystrophy (MD) is a genetic disorder that gradually weakens the body's muscles. It's caused by incorrect or missing genetic information that prevents the body from making the proteins needed to build and maintain healthy muscles.
Muscular dystrophy (MD) is a genetic disorder that gradually weakens the body's muscles. It's caused by incorrect or missing genetic information that prevents the body from making the proteins needed to build and maintain healthy muscles.  Healthy babies develop vision, movement, hearing, and other vital functions in part because enzymes clear out fatty protein and other unwanted material that can interfere with growth.
Healthy babies develop vision, movement, hearing, and other vital functions in part because enzymes clear out fatty protein and other unwanted material that can interfere with growth.